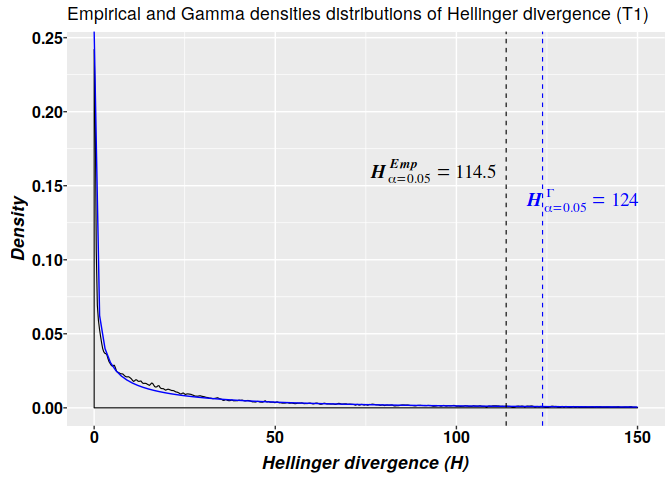
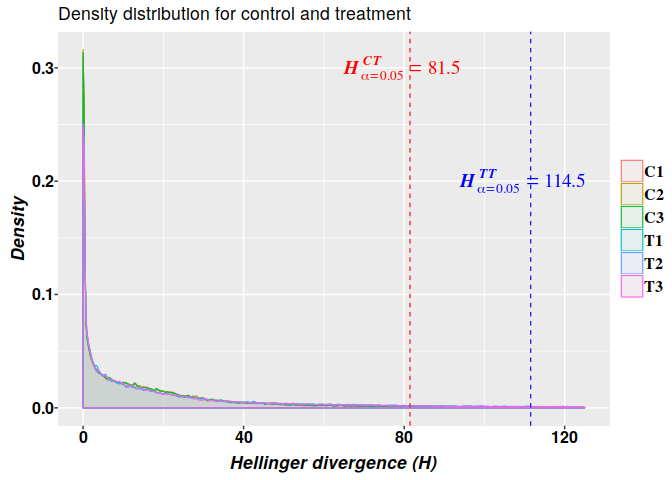
Methylation analysis with Methyl-IT is illustrated on simulated datasets of methylated and unmethylated read counts with relatively high average of methylation levels: 0.15 and 0.286 for control and treatment groups, respectively. Herein, the detection of the methylation signal is confronted as a signal detection problem. A first look on the estimation of an optimal cutoff point for the methylation signal is covered.
1. Background
Normally, there is a spontaneous variability in the control group. This is a consequence of the random fluctuations, or noise, inherent to the methylation process. The stochasticity of the the methylation process is derives from the stochasticity inherent in biochemical processes. There are fundamental physical reasons to acknowledge that biochemical processes are subject to noise and fluctuations [1,2]. So, regardless constant environment, statistically significant methylation changes can be found in control population with probability greater than zero and proportional to a Boltzmann factor [3].
Natural signals and those generated by human technology are not free of noise and, as mentioned above, the methylation signal is no exception. Only signal detection based approaches are designed to filter out the signal from the noise, in natural and in human generated signals.
The need for the application of (what is now known as) signal detection in cancer research was pointed out by Youden in the midst of the last century [4]. Here, the application of signal detection approach was performed according with the standard practice in current implementations of clinical diagnostic test [5-7]. That is, optimal cutoff values of the methylation signal were estimated on the receiver operating characteristic curves (ROCs) and applied to identify DMPs. The decision of whether a DMP detected by Fisher’s exact test (or any other statistical test implemented in Methyl-IT) is taken based on the optimal cutoff value.
2. Cutpoint for the spontaneous variability in the control group
In Methyl-IT function estimateCutPoint is used in the estimation of the optimal cutoff (cutpoint) value to distinguish signal from noise. There are also another available approaches that will be covered in a further post.
# Cutpoint estimation for FT approach
cut.ft = estimateCutPoint(LR = ft.tv, simple = TRUE,
control.names = control.nam,
treatment.names = treatment.nam,
div.col = 7L, verbose = FALSE)
# Cutpoint estimation for the FT approach using the ECDF critical value
cut.ft.hd = estimateCutPoint(LR = ft.hd, simple = TRUE,
control.names = control.nam,
treatment.names = treatment.nam,
div.col = 7L, verbose = FALSE)
cut.emd = estimateCutPoint(LR = DMP.ecdf, simple = TRUE,
control.names = control.nam,
treatment.names = treatment.nam,
div.col = 7L, verbose = FALSE)
# Cutpoint estimation for the Weibull 2-parameter distribution approach
cut.wb = estimateCutPoint(LR = DMPs.wb, simple = TRUE,
control.names = control.nam,
treatment.names = treatment.nam,
div.col = 7L, verbose = FALSE)
# Cutpoint estimation for the Gamma 2-parameter distribution approach
cut.g2p = estimateCutPoint(LR = DMPs.g2p, simple = TRUE,
control.names = control.nam,
treatment.names = treatment.nam,
div.col = 7L, verbose = FALSE)
# Control cutpoint to define TRUE negatives and TRUE positives
cuts <- data.frame(cut.ft = cut.ft$cutpoint, cut.ft.hd = cut.ft.hd$cutpoint,
cut.ecdf = cut.emd$cutpoint, cut.wb = cut.wb$cutpoint,
cut.g2p = cut.g2p$cutpoint)
cuts
## cut.ft cut.ft.hd cut.ecdf cut.wb cut.g2p ## 1 0.9847716 0.9847716 0.987013 0.988024 0.9847716
Now, with high probability true DMP can be selected with Methyl-IT function selectDIMP.
ft.DMPs <- selectDIMP(ft.tv, div.col = 7L, cutpoint = 0.9847716, absolute = TRUE)
ft.hd.DMPs <- selectDIMP(ft.hd, div.col = 7L, cutpoint = 0.9847716, absolute = TRUE)
emd.DMPs <- selectDIMP(DMP.ecdf, div.col = 7L, cutpoint = 0.9847716, absolute = TRUE)
wb.DMPs <- selectDIMP(DMPs.wb, div.col = 7L, cutpoint = 0.9847716, absolute = TRUE)
g2p.DMPs <- selectDIMP(DMPs.g2p, div.col = 7L, cutpoint = 0.9847716, absolute = TRUE)
A summary table with the number of detected DMPs by each approach:
data.frame(ft = unlist(lapply(ft.DMPs, length)), ft.hd = unlist(lapply(ft.hd.DMPs, length)),
ecdf = unlist(lapply(DMP.ecdf, length)), Weibull = unlist(lapply(wb.DMPs, length)),
Gamma = unlist(lapply(g2p.DMPs, length)))
## ft ft.hd ecdf Weibull Gamma
## C1 889 589 56 578 688
## C2 893 608 58 593 708
## C3 933 623 57 609 723
## T1 1846 1231 107 773 1087
## T2 1791 1182 112 775 1073
## T3 1771 1178 116 816 1093Nevertheless, we should evaluate the classification performance as given in the next section.
3. Evaluation of DMP classification
As shown above, DMPs are found in the control population as well. Hence, it is important to know whether a DMP is the resulting effect of the treatment or just spontaneously occurred in the control sample as well. In particular, the confrontation of this issue is extremely important when methylation analysis is intended to be used as part of a diagnostic clinical test and a decision making in biotechnology industry.
Methyl-IT function is used here, to evaluate the classification of DMPs into one of the two classes, control and treatment. Several classifiers are available to be used with this function (see the help/manual for this function or type ?evaluateDIMPclass in R console).
To evaluate the classification performances, for each methylation analysis approach, we show the results with the best available classifier. Here, the best results were found with a logistic model and a quadratic discriminant analysis (QDA) based on principal component (PC).
3.1. Evaluation of Fisher’s exact test DMP classification
ft.class = evaluateDIMPclass(LR = ft.DMPs, control.names = control.nam, treatment.names = treatment.nam, column = c(hdiv = TRUE, TV = TRUE, wprob = TRUE, pos = TRUE), classifier = "logistic", interaction = "wprob:TV", output = "conf.mat", prop = 0.6, pval.col = 11L )## Model: treat ~ hdiv + TV + logP + pos + TV:logP
## $Performance
## Confusion Matrix and Statistics
##
## Reference
## Prediction CT TT
## CT 135 29
## TT 618 1511
##
## Accuracy : 0.7178
## 95
## No Information Rate : 0.6716
## P-Value [Acc > NIR] : 1.015e-06
##
## Kappa : 0.2005
## Mcnemar's Test P-Value : < 2.2e-16
##
## Sensitivity : 0.9812
## Specificity : 0.1793
## Pos Pred Value : 0.7097
## Neg Pred Value : 0.8232
## Prevalence : 0.6716
## Detection Rate : 0.6590
## Detection Prevalence : 0.9285
## Balanced Accuracy : 0.5802
##
## 'Positive' Class : TT
##
##
## $FDR
## [1] 0.2902771
##
## $model
##
## Call: glm(formula = formula, family = binomial(link = "logit"), data = dt)
##
## Coefficients:
## (Intercept) hdiv TV logP pos
## 4.199e+01 -1.451e-01 -4.367e+01 1.092e+00 -6.602e-04
## TV:logP
## -1.546e+00
##
## Degrees of Freedom: 3437 Total (i.e. Null); 3432 Residual
## Null Deviance: 4353
## Residual Deviance: 4067 AIC: 4079
3.2. Evaluation DMP classification derived from Fisher’s exact test and ECDF critical value
ft.hd.class = evaluateDIMPclass(LR = ft.hd.DMPs, control.names = control.nam,
treatment.names = treatment.nam,
column = c(hdiv = TRUE, TV = TRUE,
wprob = TRUE, pos = TRUE),
classifier = "logistic", interaction = "wprob:TV",
pval.col = 11L, output = "conf.mat", prop = 0.6
)## Model: treat ~ hdiv + TV + logP + pos + TV:logPft.hd.class
## $Performance
## Confusion Matrix and Statistics
##
## Reference
## Prediction CT TT
## CT 174 18
## TT 325 980
##
## Accuracy : 0.7709
## 95
## No Information Rate : 0.6667
## P-Value [Acc > NIR] : < 2.2e-16
##
## Kappa : 0.3908
## Mcnemar's Test P-Value : < 2.2e-16
##
## Sensitivity : 0.9820
## Specificity : 0.3487
## Pos Pred Value : 0.7510
## Neg Pred Value : 0.9062
## Prevalence : 0.6667
## Detection Rate : 0.6546
## Detection Prevalence : 0.8717
## Balanced Accuracy : 0.6653
##
## 'Positive' Class : TT
##
##
## $FDR
## [1] 0.2490421
##
## $model
##
## Call: glm(formula = formula, family = binomial(link = "logit"), data = dt)
##
## Coefficients:
## (Intercept) hdiv TV logP pos
## 229.1917 -0.1727 -232.0026 4.4204 0.1808
## TV:logP
## -4.9700
##
## Degrees of Freedom: 2241 Total (i.e. Null); 2236 Residual
## Null Deviance: 2854
## Residual Deviance: 2610 AIC: 26223.3. Evaluation ECDF based DMP classification
ecdf.class = evaluateDIMPclass(LR = DMP.ecdf, control.names = control.nam,
treatment.names = treatment.nam,
column = c(hdiv = TRUE, TV = TRUE,
wprob = TRUE, pos = TRUE),
classifier = "pca.qda", n.pc = 4, pval.col = 10L,
center = TRUE, scale = TRUE,
output = "conf.mat", prop = 0.6
)
ecdf.class## $Performance
## Confusion Matrix and Statistics
##
## Reference
## Prediction CT TT
## CT 72 145
## TT 4 4
##
## Accuracy : 0.3378
## 95
## No Information Rate : 0.6622
## P-Value [Acc > NIR] : 1
##
## Kappa : -0.0177
## Mcnemar's Test P-Value : <2e-16
##
## Sensitivity : 0.02685
## Specificity : 0.94737
## Pos Pred Value : 0.50000
## Neg Pred Value : 0.33180
## Prevalence : 0.66222
## Detection Rate : 0.01778
## Detection Prevalence : 0.03556
## Balanced Accuracy : 0.48711
##
## 'Positive' Class : TT
##
##
## $FDR
## [1] 0.5
##
## $model
## $qda
## Call:
## qda(ind.coord, grouping = data[resp][, 1], tol = tol, method = method)
##
## Prior probabilities of groups:
## CT TT
## 0.3373134 0.6626866
##
## Group means:
## PC1 PC2 PC3 PC4
## CT 0.013261015 0.06134001 0.04739951 -0.09566950
## TT -0.006749976 -0.03122262 -0.02412678 0.04869664
##
## $pca
## Standard deviations (1, .., p=4):
## [1] 1.1635497 1.0098710 0.9744562 0.8226468
##
## Rotation (n x k) = (4 x 4):
## PC1 PC2 PC3 PC4
## hdiv -0.6692087 0.2408009 0.0214317 -0.7026488
## TV -0.3430242 -0.4422200 0.8043031 0.1996806
## logP 0.6439784 -0.1703322 0.3451435 -0.6611768
## pos -0.1406627 -0.8470202 -0.4832319 -0.1710485
##
## attr(,"class")
## [1] "pcaQDA"3.4. Evaluation of Weibull based DMP classification
ws.class = evaluateDIMPclass(LR = wb.DMPs, control.names = control.nam,
treatment.names = treatment.nam,
column = c(hdiv = TRUE, TV = TRUE,
wprob = TRUE, pos = TRUE),
classifier = "pca.qda", n.pc = 4, pval.col = 10L,
center = TRUE, scale = TRUE,
output = "conf.mat", prop = 0.6
)
ws.class
## $Performance
## Confusion Matrix and Statistics
##
## Reference
## Prediction CT TT
## CT 512 0
## TT 0 725
##
## Accuracy : 1
## 95
## No Information Rate : 0.5861
## P-Value [Acc > NIR] : < 2.2e-16
##
## Kappa : 1
## Mcnemar's Test P-Value : NA
##
## Sensitivity : 1.0000
## Specificity : 1.0000
## Pos Pred Value : 1.0000
## Neg Pred Value : 1.0000
## Prevalence : 0.5861
## Detection Rate : 0.5861
## Detection Prevalence : 0.5861
## Balanced Accuracy : 1.0000
##
## 'Positive' Class : TT
##
##
## $FDR
## [1] 0
##
## $model
## $qda
## Call:
## qda(ind.coord, grouping = data[resp][, 1], tol = tol, method = method)
##
## Prior probabilities of groups:
## CT TT
## 0.4133837 0.5866163
##
## Group means:
## PC1 PC2 PC3 PC4
## CT 0.0006129907 0.02627154 0.01436693 -0.3379167
## TT -0.0004319696 -0.01851334 -0.01012426 0.2381272
##
## $pca
## Standard deviations (1, .., p=4):
## [1] 1.3864692 1.0039443 0.9918014 0.2934775
##
## Rotation (n x k) = (4 x 4):
## PC1 PC2 PC3 PC4
## hdiv -0.70390059 -0.01853114 -0.06487288 0.7070870321
## TV -0.08754381 0.61403346 0.78440944 0.0009100769
## logP 0.70393865 0.01695660 0.06446889 0.7071255955
## pos -0.03647491 -0.78888021 0.61346321 -0.0007020890
##
## attr(,"class")
## [1] "pcaQDA"3.5. Evaluation of Gamma based DMP classification
g2p.class = evaluateDIMPclass(LR = g2p.DMPs, control.names = control.nam,
treatment.names = treatment.nam,
column = c(hdiv = TRUE, TV = TRUE,
wprob = TRUE, pos = TRUE),
classifier = "pca.qda", n.pc = 4, pval.col = 10L,
center = TRUE, scale = TRUE,
output = "conf.mat", prop = 0.6
)
g2p.class## $Performance
## Confusion Matrix and Statistics
##
## Reference
## Prediction CT TT
## CT 597 0
## TT 0 970
##
## Accuracy : 1
## 95
## No Information Rate : 0.619
## P-Value [Acc > NIR] : < 2.2e-16
##
## Kappa : 1
## Mcnemar's Test P-Value : NA
##
## Sensitivity : 1.000
## Specificity : 1.000
## Pos Pred Value : 1.000
## Neg Pred Value : 1.000
## Prevalence : 0.619
## Detection Rate : 0.619
## Detection Prevalence : 0.619
## Balanced Accuracy : 1.000
##
## 'Positive' Class : TT
##
##
## $FDR
## [1] 0
##
## $model
## $qda
## Call:
## qda(ind.coord, grouping = data[resp][, 1], tol = tol, method = method)
##
## Prior probabilities of groups:
## CT TT
## 0.3810132 0.6189868
##
## Group means:
## PC1 PC2 PC3 PC4
## CT -0.09825234 0.009217742 -0.0015298812 -0.3376429
## TT 0.06047858 -0.005673919 0.0009417082 0.2078338
##
## $pca
## Standard deviations (1, .., p=4):
## [1] 1.3934010 1.0005617 0.9910414 0.2741290
##
## Rotation (n x k) = (4 x 4):
## PC1 PC2 PC3 PC4
## hdiv -0.70097482 -0.01600430 -0.09106340 0.7071673216
## TV -0.13109213 0.26685824 0.95477779 -0.0009560507
## logP 0.70085340 0.01201179 0.09357872 0.7070383630
## pos -0.01592678 -0.96352803 0.26711394 -0.0031966410
##
## attr(,"class")
## [1] "pcaQDA"3.6. Summary of DMP classification performance
For the current simulated dataset, the best classification performance was obtained for the approach of DMP detection based on a 2-parameter gamma probability distribution model for the Hellinger divergence of methylation levels. DMPs from treatment can be distinguished from control DMPs with very high accuracy. The second best approach was obtained for the 2-parameter Weibull probability distribution model.
Obviously, for practical application, we do not need to go through all these steps. Herein, we just illustrate the need for a knowledge on the probability distributions of the signal plus noise in control and in treatment groups. The available approaches for methylation analysis are not designed to evaluate the natural variation in the control population. As a matter of fact, the phrase “natural variation” itself implies the concept of probability distribution of the variable under study. Although the probability distribution of the variable measured is objective and, as such, it does not depend on the model assumptions, the selection of the best fitted model notably improves the accuracy of our predictions.
4. Conclusions Summary
Herein, an illustrative example of methylation analysis with Methyl-IT have been presented. Whatever could be the statistical test/approach used to identify DMPs, the analysis with simulated datasets, where the average of methylation levels in the control samples is relatively high, indicates the need for the application of signal detection based approaches.
4.1. Concluding remarks
The simplest suggested steps to follow for a methylation analysis with Methyl-IT are:
- To estimate a reference virtual sample from a reference group by using function poolFromGRlist. Notice that several statistics are available to estimate the virtual samples, i.e., mean, median, sum. For experiments limited by the number of sample, at least, try the estimation of the virtual sample from the control group. Alternatively, the whole reference group can be used in pairwise comparisons with control and treatment groups (computationally expensive).
- To estimate information divergence using function estimateDivergence
- To perform the estimation of the cumulative density function of the Hellinger divergence of methylation levels using function nonlinearFitDist.
- To get the potential DMPs using function getPotentialDIMP.
- To estimate the optimal cutpoint using function estimateCutPoint.
- To retrieve DMPs with function selectDIMP.
- To evaluate the classificatio performance using function evaluateDIMPclass.
As shown here, alternative analysis is possible by using Fisher’s exact test (FT). Whether FT would be better than the approach summarized above will depend on the dataset under study. The approaches with Root Mean Square Test (RMST) and Hellinger divergence test (HDT) are also possible with function rmstGR. In these cases, we can proceed as suggested for FT. In general, RMST and HDT yield better results (not discussed here) than FT.
References
- Samoilov, Michael S., Gavin Price, and Adam P. Arkin. 2006. “From fluctuations to phenotypes: the physiology of noise.” Science’s STKE : Signal Transduction Knowledge Environment 2006 (366). doi:10.1126/stke.3662006re17.
- Eldar, Avigdor, and Michael B. Elowitz. 2010. “Functional roles for noise in genetic circuits.” Nature Publishing Group. doi:10.1038/nature09326.
- Sanchez, Robersy, and Sally A. Mackenzie. 2016. “Information Thermodynamics of Cytosine DNA Methylation.” Edited by Barbara Bardoni. PLOS ONE 11 (3). Public Library of Science: e0150427. doi:10.1371/journal.pone.0150427.
- Youden WJ. Index for rating diagnostic tests. Cancer, 1950, 3:32–5. doi:10.1002/1097-0142(1950)3:1<32::AID-CNCR2820030106>3.0.CO;2-3.
- Carter, Jane V., Jianmin Pan, Shesh N. Rai, and Susan Galandiuk. 2016. “ROC-ing along: Evaluation and interpretation of receiver operating characteristic curves.” Surgery 159 (6). Mosby: 1638–45. doi:10.1016/j.surg.2015.12.029.
- López-Ratón, Mónica, Maria Xosé Rodríguez-Álvarez, Carmen Cadarso-Suárez, Francisco Gude-Sampedro, and Others. 2014. “OptimalCutpoints: an R package for selecting optimal cutpoints in diagnostic tests.” Journal of Statistical Software 61 (8). Foundation for Open Access Statistics: 1–36. https://www.jstatsoft.org/article/view/v061i08.
- Hippenstiel, Ralph D. 2001. Detection theory: applications and digital signal processing. CRC Press.