Table of Contents
Methylation analysis with Methyl-IT is illustrated on simulated datasets of methylated and unmethylated read counts with relatively high average of methylation levels: 0.15 and 0.286 for control and treatment groups, respectively. In this part, potential differentially methylated positions are estimated following different approaches.
1. Background
Only a signal detection approach can detect with high probability real DMPs. Any statistical test (like e.g. Fisher’s exact test) not based on signal detection requires for further analysis to distinguish DMPs that naturally can occur in the control group from those DMPs induced by a treatment. The analysis here is a continuation of Part 1.
2. Potential DMPs from the methylation signal using empirical distribution
As suggested from the empirical density graphics (above), the critical values $H_{\alpha=0.05}$ and $TV_{d_{\alpha=0.05}}$ can be used as cutpoints to select potential DMPs. After setting $dist.name = “ECDF”$ and $tv.cut = 0.926$ in Methyl-IT function getPotentialDIMP, potential DMPs are estimated using the empirical cummulative distribution function (ECDF) and the critical value $TV_{d_{\alpha=0.05}}=0.926$.
DMP.ecdf <- getPotentialDIMP(LR = divs, div.col = 9L, tv.cut = 0.926, tv.col = 7, alpha = 0.05, dist.name = "ECDF")
3. Potential DMPs detected with Fisher’s exact test
In Methyl-IT Fisher’s exact test (FT) is implemented in function FisherTest. In the current case, a pairwise group application of FT to each cytosine site is performed. The differences between the group means of read counts of methylated and unmethylated cytosines at each site are used for testing (pooling.stat=”mean”). Notice that only cytosine sites with critical values $TV_d$> 0.926 are tested (tv.cut = 0.926).
ft = FisherTest(LR = divs, tv.cut = 0.926, pAdjustMethod = "BH", pooling.stat = "mean", pvalCutOff = 0.05, num.cores = 4L, verbose = FALSE, saveAll = FALSE) ft.tv <- getPotentialDIMP(LR = ft, div.col = 9L, dist.name = "None", tv.cut = 0.926, tv.col = 7, alpha = 0.05)
There is not a one-to-one mapping between $TV$ and $HD$. However, at each cytosine site $i$, these information divergences hold the inequality:
$TV(p^{tt}_i,p^{ct}_i)\leq \sqrt{2}H_d(p^{tt}_i,p^{ct}_i)$ [1].
where $H_d(p^{tt}_i,p^{ct}_i) = \sqrt{\frac{H(p^{tt}_i,p^{ct}_i)}w}$ is the Hellinger distance and $H(p^{tt}_i,p^{ct}_i)$ is given by Eq. 1 in part 1.
So, potential DMPs detected with FT can be constrained with the critical value $H^{TT}_{\alpha=0.05}\geq114.5$
4. Potential DMPs detected with Weibull 2-parameters model
Potential DMPs can be estimated using the critical values derived from the fitted Weibull 2-parameters models, which are obtained after the non-linear fit of the theoretical model on the genome-wide $HD$ values for each individual sample using Methyl-IT function nonlinearFitDist [2]. As before, only cytosine sites with critical values $TV>0.926$ are considered DMPs. Notice that, it is always possible to use any other values of $HD$ and $TV$ as critical values, but whatever could be the value it will affect the final accuracy of the classification performance of DMPs into two groups, DMPs from control and DNPs from treatment (see below). So, it is important to do an good choices of the critical values.
nlms.wb <- nonlinearFitDist(divs, column = 9L, verbose = FALSE, num.cores = 6L)
# Potential DMPs from 'Weibull2P' model
DMPs.wb <- getPotentialDIMP(LR = divs, nlms = nlms.wb, div.col = 9L,
tv.cut = 0.926, tv.col = 7, alpha = 0.05,
dist.name = "Weibull2P")
nlms.wb$T1 ## Estimate Std. Error t value Pr(>|t|)) Adj.R.Square
## shape 0.5413711 0.0003964435 1365.570 0 0.991666592250838
## scale 19.4097502 0.0155797315 1245.833 0
## rho R.Cross.val DEV
## shape 0.991666258901194 0.996595712743823 34.7217494754823
## scale
## AIC BIC COV.shape COV.scale
## shape -221720.747067975 -221694.287733122 1.571674e-07 -1.165129e-06
## scale -1.165129e-06 2.427280e-04
## COV.mu n
## shape NA 50000
## scale NA 500005. Potential DMPs detected with Gamma 2-parameters model
As in the case of Weibull 2-parameters model, potential DMPs can be estimated using the critical values derived from the fitted Gamma 2-parameters models and only cytosine sites with critical values $TV_d > 0.926$ are considered DMPs.
nlms.g2p <- nonlinearFitDist(divs, column = 9L, verbose = FALSE, num.cores = 6L,
dist.name = "Gamma2P")
# Potential DMPs from 'Gamma2P' model
DMPs.g2p <- getPotentialDIMP(LR = divs, nlms = nlms.g2p, div.col = 9L,
tv.cut = 0.926, tv.col = 7, alpha = 0.05,
dist.name = "Gamma2P")
nlms.g2p$T1## Estimate Std. Error t value Pr(>|t|)) Adj.R.Square
## shape 0.3866249 0.0001480347 2611.717 0 0.999998194156282
## scale 76.1580083 0.0642929555 1184.547 0
## rho R.Cross.val DEV
## shape 0.999998194084045 0.998331895911125 0.00752417919133131
## scale
## AIC BIC COV.alpha COV.scale
## shape -265404.29138371 -265369.012270572 2.191429e-08 -8.581717e-06
## scale -8.581717e-06 4.133584e-03
## COV.mu df
## shape NA 49998
## scale NA 49998Summary table:
data.frame(ft = unlist(lapply(ft, length)), ft.hd = unlist(lapply(ft.hd, length)),
ecdf = unlist(lapply(DMPs.hd, length)), Weibull = unlist(lapply(DMPs.wb, length)),
Gamma = unlist(lapply(DMPs.g2p, length)))## ft ft.hd ecdf Weibull Gamma
## C1 1253 773 63 756 935
## C2 1221 776 62 755 925
## C3 1280 786 64 768 947
## T1 2504 1554 126 924 1346
## T2 2464 1532 124 942 1379
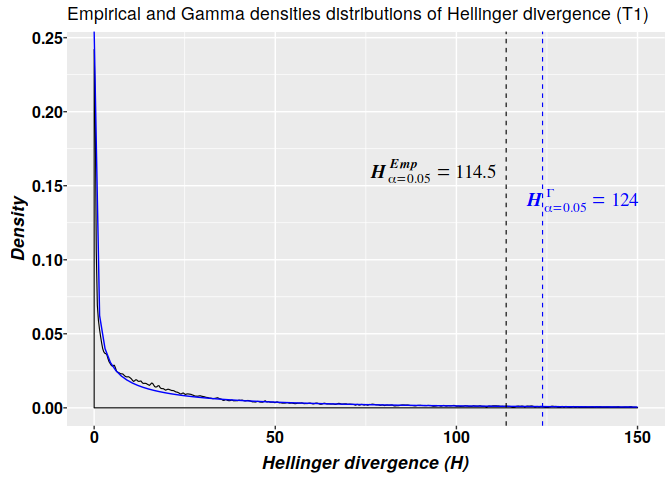
## T3 2408 1477 121 979 1354The graphics for the empirical (in black) and Gamma (in blue) densities distributions of Hellinger divergence of methylation levels for sample T1 are shown below. The 2-parameter gamma model is build by using the parameters estimated in the non-linear fit of $H$ values from sample T1. The critical values estimated from the 2-parameter gamma distribution $H^{\Gamma}_{\alpha=0.05}=124$ is more ‘conservative’ than the critical value based on the empirical distribution $H^{Emp}_{\alpha=0.05}=114.5$. That is, in accordance with the empirical distribution, for a methylation change to be considered a signal its $H$ value must be $H\geq114.5$, while according with the 2-parameter gamma model any cytosine carrying a signal must hold $H\geq124$.
suppressMessages(library(ggplot2)) # Some information for graphic dt <- data[data$sample == "T1", ] coef <- nlms.g2p$T1$Estimate # Coefficients from the non-linear fit dgamma2p <- function(x) dgamma(x, shape = coef[1], scale = coef[2]) qgamma2p <- function(x) qgamma(x, shape = coef[1], scale = coef[2]) # 95 q95 <- qgamma2p(0.95) # Gamma model based quantile emp.q95 = quantile(divs$T1$hdiv, 0.95) # Empirical quantile # Density plot with ggplot ggplot(dt, aes(x = HD)) + geom_density(alpha = 0.05, bw = 0.2, position = "identity", na.rm = TRUE, size = 0.4) + xlim(c(0, 150)) + stat_function(fun = dgamma2p, colour = "blue") + xlab(expression(bolditalic("Hellinger divergence (HD)"))) + ylab(expression(bolditalic("Density"))) + ggtitle("Empirical and Gamma densities distributions of Hellinger divergence (T1)") + geom_vline(xintercept = emp.q95, color = "black", linetype = "dashed", size = 0.4) + annotate(geom = "text", x = emp.q95 - 20, y = 0.16, size = 5, label = 'bolditalic(HD[alpha == 0.05]^Emp==114.5)', family = "serif", color = "black", parse = TRUE) + geom_vline(xintercept = q95, color = "blue", linetype = "dashed", size = 0.4) + annotate(geom = "text", x = q95 + 9, y = 0.14, size = 5, label = 'bolditalic(HD[alpha == 0.05]^Gamma==124)', family = "serif", color = "blue", parse = TRUE) + theme( axis.text.x = element_text( face = "bold", size = 12, color="black", margin = margin(1,0,1,0, unit = "pt" )), axis.text.y = element_text( face = "bold", size = 12, color="black", margin = margin( 0,0.1,0,0, unit = "mm")), axis.title.x = element_text(face = "bold", size = 13, color="black", vjust = 0 ), axis.title.y = element_text(face = "bold", size = 13, color="black", vjust = 0 ), legend.title = element_blank(), legend.margin = margin(c(0.3, 0.3, 0.3, 0.3), unit = 'mm'), legend.box.spacing = unit(0.5, "lines"), legend.text = element_text(face = "bold", size = 12, family = "serif")
- Steerneman, Ton, K. Behnen, G. Neuhaus, Julius R. Blum, Pramod K. Pathak, Wassily Hoeffding, J. Wolfowitz, et al. 1983. “On the total variation and Hellinger distance between signed measures; an application to product measures.” Proceedings of the American Mathematical Society 88 (4). Springer-Verlag, Berlin-New York: 684–84. doi:10.1090/S0002-9939-1983-0702299-0.
- Sanchez, Robersy, and Sally A. Mackenzie. 2016. “Information Thermodynamics of Cytosine DNA Methylation.” Edited by Barbara Bardoni. PLOS ONE 11 (3). Public Library of Science: e0150427. doi:10.1371/journal.pone.0150427.